

ADESÃO E AGREGAÇÃO PLAQUETÁRIA
O princípio da Hemostasia
As plaquetas são fragmentos do citoplasma dos megacariócitos e apresentam meia-vida de 10 dias. Desempenham um papel crucial no sistema hemostático, trabalhando incessantemente para manter o equilíbrio entre hemorragia e trombose. A adesão e agregação plaquetária são processos intrincados que garantem a formação de um coágulo eficaz quando necessário.
A principal função das plaquetas é a formação do tampão plaquetário, que interrompe temporariamente o sangramento após uma lesão vascular. Além disso, exercem também ação pró-coagulante, por meio da interação dos fatores da coagulação com receptores específicos localizados na superfície plaquetária.
Adesão Plaquetária
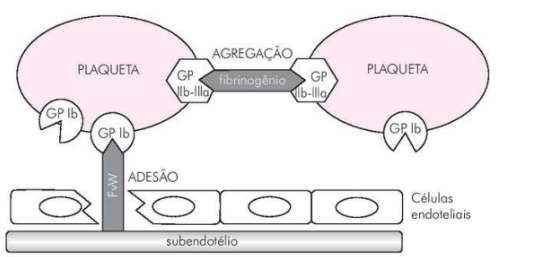
Quando ocorre uma lesão em um vaso, a exposição do subendotélio vascular desencadeia uma série de eventos complexos. A adesão plaquetária é o primeiro passo desse processo. As plaquetas, anteriormente inertes, reconhecem e aderem às moléculas de matriz extracelular expostas, se ligam ao colágeno do subendotélio através da glicoproteína lb-lX presente em sua membrana. Essa ligação é mediada pelo fator de von Willebrand.
Agregação Plaquetária
Após a adesão, as plaquetas ativadas liberam uma série de mediadores químicos, incluindo o tromboxano A2, ADP (difosfato de adenosina) e trombina. Esses mediadores desempenham um papel central na ativação e recrutamento de plaquetas adicionais para o local da lesão, promovendo a agregação plaquetária.
O ADP age nos receptores de P2Y1 e P2Y12 nas membranas plaquetárias, desencadeando uma série de eventos intracelulares que levam à expressão do complexo glicoproteico IIb/IIIa na superfície das plaquetas. O complexo IIb/IIIa é essencial para a ligação entre as plaquetas, pois serve como receptor para fibrinogênio e outros ligantes, facilitando a formação de agregados plaquetários.
A ativação do complexo IIb/IIIa e a ligação do fibrinogênio resultam na formação de pontes entre as plaquetas, levando à formação de um tampão plaquetário no local da lesão. Esse tampão reduz o fluxo sanguíneo através do vaso danificado e fornece uma superfície adequada para a coagulação sanguínea subsequente.
Avaliação laboratorial
A avaliação da função plaquetária se dá através de exames que avaliam tanto a morfologia como a quantidade desses fragmentos celulares. No plaquetograma conseguimos apresenta parâmetros plaquetários como volume (VPM – Volume Corpuscular Médio) e distribuição (PDW – índice de anisocitose plaquetária); além do valor quantitativo de plaquetas que deve variar entre 150.000 mm³ e 450.000 mm³. Saiba mais sobre o plaquetograma em: https://inml.com.br/plaquetograma/
Patologias Associadas
Por ter diversos mecanismos de ativação e inibição, a adesão e agregação plaquetária podem sofrer alterações em sua fisiologia normal através de diversas patologias, incluindo algumas condições genéticas como:
- Doença de von Willebrand
A doença de von Willebrand é a coagulopatia hereditária mais comum, causada por mutações no gene que codifica o fator de von Willebrand (FvW), impedindo que a adesão plaquetária ocorra de maneira eficaz. Os sintomas hemorrágicos resultam da disfunção plaquetária ou atividade reduzida do fator VIII. Na avaliação laboratorial, geralmente há plaquetometria normal, tempo de sangramento prolongado e TTPA alterado em alguns casos, enquanto o TP permanece normal.
- Síndrome de Bernard Soulier
Esta condição rara, de origem genética e recessiva, caracteriza-se por disfunção plaquetária, redução do número de plaquetas e propensão a sangramentos. A base molecular da doença consiste na deficiência genética da glicoproteína Ib-IX, que atua como receptor do FvW na membrana plaquetária. Isso prejudica significativamente o mecanismo de adesão plaquetária, afetando testes como o tempo de sangramento e a agregação plaquetária.
- Púrpura trombocitopênica imunológica (PTI)
A PTI é uma condição caracterizada pela produção de autoanticorpos direcionados contra as plaquetas, os quais desencadeiam sua destruição no sistema reticuloendotelial, especialmente no baço, resultando em uma diminuição significativa de sua vida útil na circulação. Embora a destruição periférica das plaquetas seja o mecanismo principal, a incapacidade dos megacariócitos na medula óssea de aumentar a produção de plaquetas contribui para o quadro clínico. Saiba mais sobre a PTI aqui: https://inml.com.br/purpura-trombocitopenica-idiopatica/
Em conclusão, a adesão e agregação plaquetária representam processos cruciais no sistema hemostático, desempenhando um papel fundamental na prevenção de hemorragias excessivas e na formação de coágulos sanguíneos após lesões vasculares. A compreensão desses mecanismos complexos e suas patologias associadas é essencial para o desenvolvimento de novas estratégias terapêuticas, bem como para o diagnóstico e tratamento de distúrbios hemostáticos e cardiovasculares.
O próximo passo de todo analista que deseja ter mais segurança na bancada
Todo analista que busca se destacar e se tornar um profissional mais atualizado, capacitado e qualificado para o mercado de trabalho precisa considerar uma pós-graduação.
Um profissional com especialização é valorizado na área laboratorial; esse é um fato inegável.
Unimos o útil ao agradável ao desenvolver uma pós-graduação em Hematologia Laboratorial e Clínica.
Para aqueles que procuram a comodidade de uma pós-graduação 100% online e ao vivo, sem abrir mão da excelência no ensino, temos a solução ideal.
Nossa metodologia combina teoria e prática da rotina laboratorial, garantindo um aprendizado efetivo.
Contamos com um corpo docente altamente qualificado, com os melhores professores do Brasil, referências em suas áreas de atuação.
No Instituto Nacional de Medicina Laboratorial, temos apenas um objetivo: mais do que ensinar, vamos tornar VOCÊ uma referência.
Toque no botão abaixo e conheça a pós-graduação em Hematologia Laboratorial e Clínica.
QUERO CONHECER TODOS OS DETALHES DA PÓS-GRADUAÇÃO
Referências
Atlas de hematologia : clínica hematológica ilustrada/ Therezinha Ferreira Lorenzi, coordenadora. – Rio de Janeiro: Guanabara Koogan, 2006
JOÃO, Cristina. Doença de von Willebrand. Medicina Interna, v. 8, n. 1, p. 28-36, 2001.
Naoum, Flávio Augusto Doenças que alteram os exames hematológicos / Flávio Augusto Naoum. – 2. ed. -Rio de Janeiro: Atheneu, 2017

